Research Topics¶
Introduction¶
In molecular assemblies, diverse properties emerge as many molecules with distinct characteristics interact with each other. Understanding how collective behavior emerges from individual molecular properties can provide valuable insights for various fields, including materials science, nanotechnology, and pharmaceuticals.
Molecular simulation reproduces materials as assemblies of molecules on a computer, allowing us to actually observe the arrangement and motion of molecules. Moreover, since information about the reproduced material exists within the computer, we can investigate phenomena more deeply than by experimental means alone. Thus, the molecular simulation provides a powerful means of understanding phenomena in conjunction with experiments and theories.
This page introduces some of our current research and past achievements.
Elucidating Mass Transport in Heterogeneous Systems¶
Development and Application of Multiscale Computational Methods Based on Free-Energy Landscape and Position-Dependent Diffusion ¶
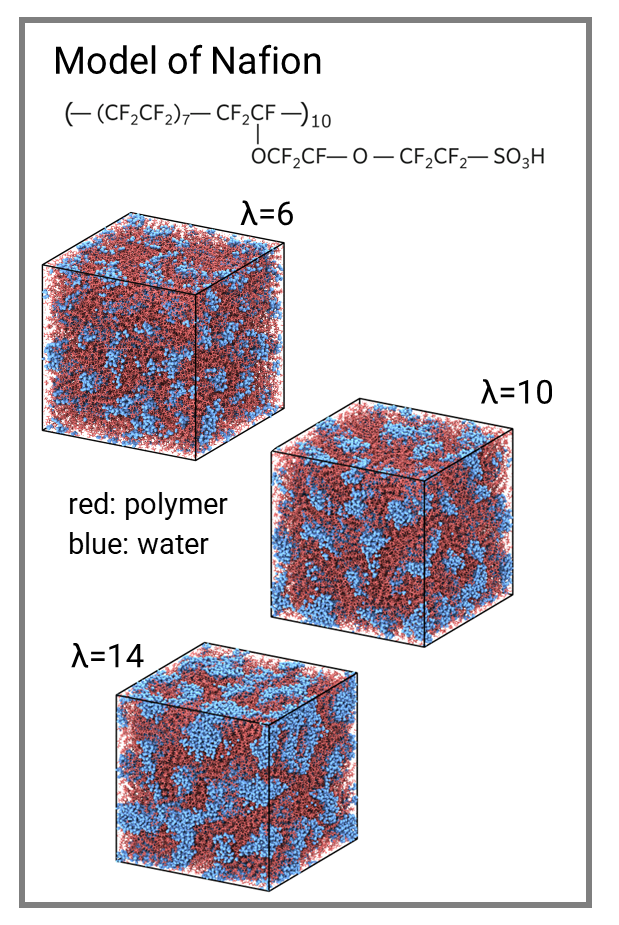
Numerous materials exhibit nanoscale heterogeneity, and the mass transport occurring within them is of interest across various fields. In particular, we have worked on mass transport related to fuel cells.
Polymer electrolyte membranes for fuel cells, such as Nafion, are highly heterogeneous. The polymer has a hydrophobic main chain and hydrophilic side chains with sulfonate groups. Because the water only mixes with the latter, the microphase separation occurs at a scale of approximately 3-5 nm (see Figure 1). Additionally, porous carbon used in electrode catalyst layers is another example of heterogeneous systems, where nanometer-scale pores are intentionally formed in the material. During fuel cell operation, transport of oxygen, hydrogen, ions, and water occurs in such heterogeneous environments. Elucidating transport mechanisms and subsequent rational material design are critical challenges for improving fuel cell performance.
In mass transport within heterogeneous systems, transport pathways wind, split, and merge repeatedly. In addition, distinct local states exist along the pathways and thus both mobility and accessibility can vary along the pathways. However, experimental quantification of such local information is unrealistic. Furthermore, mass transport in heterogeneous systems is challenging for conventional molecular dynamics simulations because of the timescales involved.
Therefore, we have developed a multiscale computational approach in which we determine the free-energy landscape , which is a measure of the driving force acting on transported molecules, and the position-dependent diffusion constant , which is a measure of mobility, with angstrom-scale resolution using molecular dynamics calculations. We then use these physical quantities as input fields to perform dynamic Monte Carlo simulations that satisfy the diffusion equation. Additionally, we are developing a method for finding plausible pathways once and are obtained.
Recently, as part of a program for promoting research on the supercomputer Fugaku, we have developed a machine learning model that predicts the free-energy landscape and position-dependent diffusion constant . Using this machine learning model, we are working with other collaborators on mass transport in fuel cell cathode catalyst layers (see Figure 2).
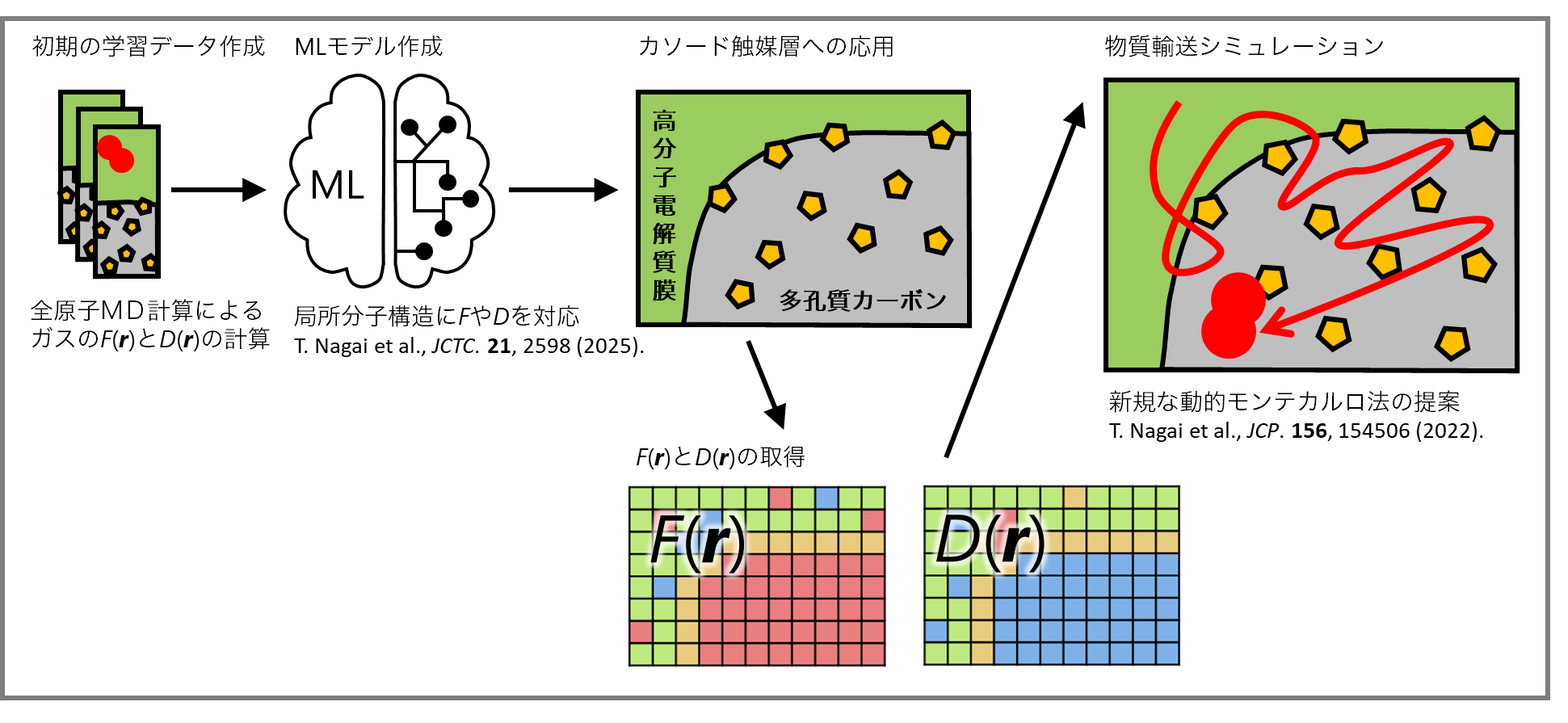
Keywords¶
Polymer Electrolyte Membranes, Free-Energy Landscape, Position-Dependent Diffusion Constant, Dynamic Monte Carlo, Machine Learning Model
Related Publications¶
-
T. Nagai, Nobuaki Kikkawa, Ryosuke Jinnouchi, Masayuki Kimura, and Susumu Okazaki, Journal of Chemical Theory and Computation 21, 2598–2611 (2025); https://doi.org/10.1021/acs.jctc.4c01552.
-
T. Nagai and Koji Yoshida, Journal of the Physical Society of Japan 93, 084805 (7 pages) (2024); https://doi.org/10.7566/JPSJ.93.084805.
-
Yoshimichi Andoh, Shin-ichi Ichikawa, Tatsuya Sakashita, Kazushi Fujimoto, Noriyuki Yoshii, T. Nagai, Zhiye Tang, and Susumu Okazaki, The Journal of Chemical Physics 158, 194803 (12 pages) (2023); https://doi.org/10.1063/5.0144361.
-
T. Nagai and Susumu Okazaki, The Journal of Chemical Physics 157, 054502 (10 pages) (2022); https://doi.org/10.1063/5.0096574.
-
T. Nagai, Akira Yoshimori, and Susumu Okazaki, The Journal of Chemical Physics 156, 154506 (14 pages) (2022); https://doi.org/10.1063/5.0086949.
-
T. Nagai, Kazushi Fujimoto, and Susumu Okazaki, The Journal of Chemical Physics 156, 044507 (14 pages) (2022); https://doi.org/10.1063/5.0075969.
-
T. Nagai, Shuhei Tsurumaki, Ryo Urano, Kazushi Fujimoto, Wataru Shinoda, and Susumu Okazaki, Journal of Chemical Theory and Computation 16, 7239–7254 (2020); https://doi.org/10.1021/acs.jctc.0c00448.